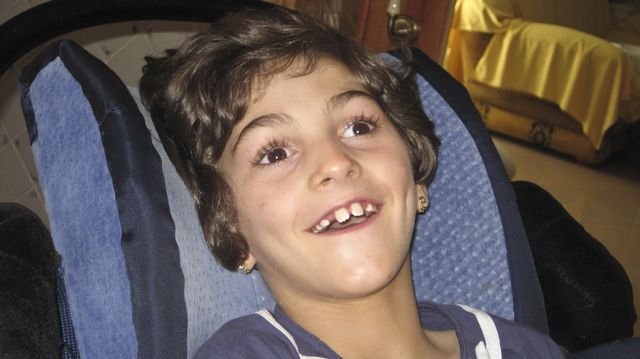
Un grupo de investigadores españoles ha descubierto un nuevo síndrome neurodegenerativo infantil, conocido como Encefalopatía de Celia , en memoria de Celia Carrión Perez de Tudela, hija de Naca Perez de Tudela (Presidenta de la Asociación de enfermedades raras Dgenes y de la Asociación de familiares y afectados de lipodistrofias AELIP ) y Juan Carrion (Presidente de la Federación Española de enfermedades raras FEDER). El trabajo acaba de ser publicado en la versión digital del Journal of Medical Genetics.
David Araújo-Vilar y Jesús Requena, de la Universidad de Santiago de Compostela (USC), en colaboración con dos expertas del Hospital Clínico Universitario Virgen de la Arrixaca de Murcia las doctoras Encarna Guillen y Rosario Domingo , han descrito un nuevo síndrome neurodegenerativo infantil junto con sus bases genéticas y moleculares.
Esta nueva enfermedad –denominada Encefalopatía de Celia en memoria del primer caso reconocido– se inicia en la primera infancia como un retraso psicomotriz, y a partir de los tres o cuatro años los niños sufren un proceso neurodegenerativo que los lleva a la muerte antes de los nueve.
CELIA TE DA LAS GRACIAS .
Tu generosidad y colaboración permite a la ciencia descubrir un nuevo dia para seguir disfrutando de la vida. VISITA Y COLABORA www.aelip.org Contacta con AELIP: info@aelip.org
Según los autores del trabajo, publicado en la versión on line del Journal of Medical Genetics, “se trata de una enfermedad autosómica recesiva, con la circunstancia particular de que los pacientes heterocigotos compuestos presentaron, además del cuadro neurológico, lipodistrofia generalizada”.
La dolencia está producida por una mutación hasta ahora no conocida en el gen BSCL2 que codifica para la proteína seipina. Esta nueva mutación da lugar a una región de ‘splicing’ alternativo que conduce a la pérdida del exón 7 del gen, provocando así una proteína aberrante que se expresa hasta 1.000 veces más en el cerebro de los enfermos.
Los estudios anatomo-patológicos de dos pacientes pusieron de manifiesto una marcada afectación de la corteza cerebral y los ganglios de la base, con unas manifestaciones inmunohistoquímicas que se asemejan a las observadas en la enfermedad de Huntington.
Los pacientes identificados –un total de seis, de los que cinco ya han fallecido– son originarios de tres municipios Jumilla, Mula y Totana , de la comunidad autónoma de la Region de Murcia.
El descubrimiento de las bases geneticas y moleculares de esta nueva enfermedad , es un paso muy importante, desde el punto de vista cientifico y tambien para la clinica. A partir de ahora, sera mas facil el dioagnostico de otros casos y las familias em ñas que se detecte esta alteración pdoran recibir consejo genetico y acogerse al diagnostico preimplantacional,.
Referencia bibliográfica
Encarna Guillén-Navarro, Sofía Sánchez-Iglesias, Rosario Domingo-Jiménez, Berta Victoria, Alejandro Ruiz-Riquelme, Alberto Rábano, Lourdes Loidi, Andrés Beiras, Blanca González-Méndez, Adriana Ramos, Vanesa López-González, María Juliana Ballesta-Martínez, Miguel Garrido-Pumar, Pablo Aguiar, Alvaro Ruibal, Jesús R Requena, and David Araújo-Vilar. A new seipin-associated neurodegenerative syndrome. J Med Genet jmedgenet-2013-101525 Published Online First: 6 April 2013 doi:10.1136/jmedgenet-2013-101525 Journal of Medical Genetics.